Author: Hsiangkuo Yuan 1, Anna Y. Chen 2, Stephen D. Silberstein * 3
Author Affiliation:
1 Department of Neurology, Thomas Jefferson University, Philadelphia, PA, USA
2 Sidney Kimmel Medical College, Thomas Jefferson University, Philadelphia, PA, USA
3 Jefferson Headache Center, Thomas Jefferson University, Philadelphia, PA, USA
Research Support: Support was provided solely from institutional and/or departmental sources.
Competing Interests: The author/s declare no competing interest.
Issue: 01.03
DOI: doi.org/10.30756/ahmj.2020.01.03
Received Date: Dec 2, 2019
Accepted Date: Dec 16, 2019
Published Date: January 2, 2020
Recommended Citation: Yuan H, Chen AY, Silberstein SD. CGRP Therapeutics For The Treatment Of Migraine – A Narrative Review. Ann Head Med. 2020;01:03. DOI: 10.30756/ahmj.2020.01.03
Abstract
Migraine is a highly prevalent, disabling, and costly primary headache disorder whose pathogenesis involves the dysfunctional activation of the trigeminovascular system. Calcitonin gene-related peptide (CGRP) is the most prevalent neuropeptide released by activation of trigeminal afferents and is considered to play a major role in pain sensitization within the trigeminovascular system. Novel therapeutic agents have been developed to prevent and treat migraine by targeting and antagonizing CGRP functions. These agents consist of gepants, which are small molecule CGRP antagonists, and monoclonal antibodies (mAbs) that target either the CGRP ligands or receptors. There are currently 4 second-generation gepants being studied: ubrogepant, rimegepant, and vazegepant for the treatment of acute migraine attacks, and atogepant for migraine prevention. CGRP mAbs are currently indicated for migraine prevention; the three FDA-approved mAbs for migraine prevention are erenumab, galcanezumab, and fremanezumab. Eptinezumab is currently under FDA review. Gepants and CGRP mAbs are effective and safe in the treatment of migraine. CGRP mAbs even work among those who failed multiple other preventive medications. These medications are emerging as promising treatment options for migraine patients.
Introduction
Migraine is a common primary headache disorder affecting more than a billion people worldwide. It has been ranked as the second-highest cause of years lived with disability by the World Health Organization (WHO).1 The projected financial burden of migraine in the United States is estimated to be about US $11 billion.2 Migraine attacks typically last between 4-72 hours. Common migraine characteristics include unilateral location, pulsating quality, aggravation by routine physical activity, and association with nausea, vomiting, and/or photophobia and phonophobia. Migraine can be classified as with aura or without aura. An aura is a complex of fully reversible visual, sensory, or other central nervous system symptoms that usually occur before the onset of headache but may also begin after the headache has started or continue into the headache phase. Visual aura is the most common type and is present in 90% of patients with migraine with aura, but sensory and speech/motor disturbances may also occur. In addition to aura, migraine is often accompanied by a nonpainful prodrome and postdrome. Prodrome (premonitory) symptoms such as fatigue/cognitive change, homeostatic alterations, and sensory sensitivities (e.g., nausea, photophobia, phonophobia) can start hours to days before the headache phase with possible involvement in the hypothalamus, brainstem, and some cortical areas.3 Postdrome symptoms (neuropsychiatric, sensory, gastrointestinal, general symptoms, etc.) also can last for several hours after the headache resolves.4 Based on the attack frequency, migraine can be classified as chronic (³ 15 headache days per month) or episodic (< 15 headache days per month).5 It is estimated that 2.5% of episodic migraine (EM) patients progress yearly to chronic migraine (CM), which affects roughly 1-3% of population. The risk factors of migraine chronification include acute medication overuse, depression, anxiety, poor sleep, caffeine overuse, stress, obesity, noncephalic pain, head/neck injury, allodynia, and poor self-efficacy.6 With such a profound socioeconomical impact, there is a strong need to further understand the pathophysiology of migraine and develop new migraine-specific treatments.
Great challenges remain in managing migraine. Many clinically effective migraine medications, FDA-approved or off-label, display unique adverse event (AE) profiles that influence medication compliance. It has been shown that adherence to preventive medication drops significantly after 30 days with only 25% adherence at 6 months,7 mainly due to drug AEs and poor efficacy. As of May 2018, the FDA has approved six oral preventives for migraine, one injectable preventive specifically for CM, and dihydroergotamine and several triptans for acute treatment. Recently, calcitonin-gene related peptide (CGRP) antagonizing therapy has emerged as an effective preventive medication with minimal AEs. At the time of writing, the FDA has approved 3 CGRP-targeting monoclonal antibodies (mAbs): erenumab (Aimovig; Amgen, Thousand Oaks, CA), fremanezumab (Ajovy; TEVA, Petah Tikva, Israel), and galcanezumab (Emgality; Eli Lilly, Indianopolis, IN). There is also another CGRP-targeting mAb, eptinezumab (Lundbeck, Deerfield, IL), and 3 CGRP antagonizing small molecules (gepants), namely ubrogepant (Allergan, Dublin, Ireland), atogepant (Allergan, Dublin, Ireland), and rimegepant (New Haven, CT) currently under FDA review. In this article, we discuss the role of CGRP in migraine pathophysiology and compare the efficacy of different CGRP antagonizing modalities available to date.
Migraine Pathophysiology
The pathophysiology of migraine involves a dysfunctional activation of the trigeminovascular nociceptive system leading to the sensation of head pain.8 Trigeminal afferents are pseudo-unipolar neurons whose cell bodies form the trigeminal ganglion (TG) and interact with the surrounding satellite glial cells and other cell types. Together with afferents from the upper cervical cord, they innervate pain-sensitive structures in the head and communicate with the trigeminal cervical complex (TCC), then project to multiple central nuclei (e.g., thalamus, hypothalamus, brainstem, basal ganglia, etc.) that are involved in processing head pain as well as other migraine-associated symptoms. Activation of the trigeminovascular nociceptive system in migraine causes neurogenic inflammation, and also leads to pain sensitization both peripherally and centrally with subsequent sustained headache.9 Trigeminal afferent activation releases a variety of neuropeptides, including CGRP, substance P, and others, with CGRP being the most abundant.10, 11, 12
Neuropeptides play a major role in pain signaling and modulation.13 Unlike neurotransmitters which are synthesized at the presynaptic terminals, neuropeptides are synthesized by the rough endoplasmic reticulum, loaded into dense core vesicles (DCVs), transported to the distant varicosities along the axon, and released via exocytosis. Different neuropeptides often colocalize in the same DCV. These peptides are released mostly from nonsynaptic sites and act locally on nerve fibers or cells within a few microns nearby.14 Since there is no known reuptake system for neuropeptides, they are degraded by mast cell tryptase, neutral endopeptidase and matrix metalloproteinase II.15 Most neuropeptides act by binding to their corresponding G protein-coupled receptors (GPCRs) that are usually heterogeneously distributed throughout the nervous system. In contrast to neurotransmitters that act on fast ion channels, the neuropeptide receptive nerve fibers or cells typically respond in seconds to minutes thereby modulating the neuronal or glial activities. The exact roles of these neuropeptides, albeit not fully known, are becoming more understood.
CGRP is a 37-amino acid neuropeptide broadly distributed in both neuronal and non-neuronal regions throughout the body. CGRP belongs to the calcitonin family of peptides, which include calcitonin, amylin, adrenomedullin, and adrenomedullin-2.16 There are two isoforms of CGRP, a-CGRP and b-CGRP, that are encoded by two different genes and differ in three amino acids.17, 18 CGRP serves many purposes throughout the body, including sensory, digestive, vascular, vestibular, hematopoietic, immunomodulatory, nociceptive, and tissue healing functions.19 It is the most potent vasodilator currently known, and has been shown to have a potency that is ~10-fold higher than most potent prostaglandins and 10-100 times greater than other vasodilators such as acetylcholine and substance P.20 In particular, CGRP is the most abundant neuropeptide in the trigeminal system. It is found mainly in small capsaicin-sensitive C fibers that follow the cerebral and meningeal arteries and innervate pain-sensitive structures; no CGRP is found in the second-order trigeminal neurons. CGRP is not permeable across the blood brain barrier; it acts on nearby receptive cells and can diffuse beyond the release site via volume transmission. Unmetabolized intracranial CGRP may re-enter circulation but exhibits a rather short serum half-life (~7 minutes).21 Activation of trigeminal nociceptive fibers results in antidromic release of CGRP, which binds to both the canonical CGRP receptor and the amylin 1 (AMY1) receptor on the neurons, nodes of Ranvier, glia, blood vessel smooth muscles, immune cells, and others. The CGRP receptor is a GPCR that requires the heterodimerization of 2 components (calcitonin receptor-like receptor and a single transmembrane-spanning protein called receptor activity-modifying protein 1) that is then coupled to receptor component protein for effective CGRP signaling. Upon activation, the adenylyl cyclase catalyzes the synthesis of cAMP along with other subsequent intracellular messengers generating a variety of functions depending on the cell type.22, 23
CGRP’s Role In Migraine
CGRP was first demonstrated to play a role in migraine in 1990, when it was revealed that CGRP levels were increased in the jugular venous outflow following triggered migraine attacks.3 CGRP level increase was later confirmed during spontaneous migraine attacks; treatment with triptans normalized headaches and CGRP levels.24, 25, 26 Such normalization was also observed in subjects who received topiramate and onabotulinumtoxin A;27, 28 onabotulinumtoxin A directly inhibits CGRP release from C-fibers,29 and the 5-HT1B/1D/1F receptors found on CGRP-containing trigeminal neurons that are targeted by triptans also inhibit the release of CGRP.30, 31 In addition, CGRP infusion triggers delayed migraine-like attacks in roughly 68% of migraine subjects but not in healthy controls or individuals with tension-type headaches or familial hemiplegic migraine.32, 33, 34, 35 This highly suggests CGRP’s pathogenic role in migraine but the exact migraine triggering mechanism remains unknown. In animal models, even at vasodilatory doses, neither intravenous (IV) nor topical dural application of CGRP sensitized meningeal nociceptors.36 Since the vascular theory of migraine is no longer favored, it has been theorized that CGRP mediates neurogenic inflammation and modulates nociceptive inputs that contribute to migraine.37 CGRP and nitric oxide (NO) from activated TG stimulate adjacent satellite glial cells to release IL-1b, IL-6, and other cytokines and increase cyclooxygenase activity and PGE2 production.38, 39, 40, 41 Since glial inhibition reversed CGRP-induced thermal nociception,38 neuro-glial crosstalk likely maintains a state of sensitization interaction. These studies suggest that CGRP, rather than eliciting a direct excitation effect, likely promotes and maintains pain sensitization. Also, CGRP serum levels were found to be elevated after NO donor administration to sumatriptan-sensitized rather than non-sensitized rats.42 These findings may explain why CGRP infusion triggered a delayed headache only in subjects with migraine. The exact CGRP mechanism of action is likely multifactorial within the trigeminovascular nociceptive system but remains to be explored. Nonetheless, disrupting this sensitization process by blocking the CGRP function appears to be clinically relevant in migraine. To date, there are 2 types of CGRP functional blocking agents: small molecule CGRP antagonists known as gepants and CGRP targeting mAbs.
CGRP Antagonists – Gepants
CGRP antagonists are small molecules known as “gepants” that were developed in the 1990s for the treatment of migraine. They bind to CGRP receptors and can reverse CGRP-induced vasodilation as well as neurogenic inflammation that leads to migraine. Olcegepant (Boehringer Ingelheim GmbH, Germany), telcagepant (Merck, Kenilworth, NJ) and MK-3207 (Merck, Kenilworth, NJ) are early gepants whose efficacy were found to be better than placebo and comparable to that of triptans.43, 44, 45, 46 However, further development of these first-generation gepants was halted by their potential hepatotoxic effects when dosed regularly.47 Currently, second-generation gepants are being developed with no major hepatotoxicity seen so far. The 4 gepants in this generation are ubrogepant (Allergan, Dublin, Ireland), atogepant (Allergan, Dublin, Ireland), rimegepant (Biohaven, New Haven, CT), and vazegepant (Biohaven, New Haven, CT). Gepants are competitive receptor antagonists. They bind to CGRP receptors, block cAMP production, and inhibit trigeminovascular nociceptive activation.48 In animal studies, IV olcegepant pretreatment inhibited capsaicin-induced Fos expression in the spinal trigeminal nucleus but not in the TG.49 Olcegepant blocks cAMP response element-binding protein (CREB) phosphorylation more potently than cAMP accumulation in TG neurons, especially those with AMY1 receptors.50 These features may be unique to olcegepant, but TG neurons likely are a target of interest. Satellite glial cells, the predominant cell type in TG, express CGRP receptors and may also be another target. However, TG-derived glia cells have a weaker response to CGRP,50, 51 and may react differently to CGRP antagonism. The effect of CGRP antagonism on other cell types in TG remains unclear. In a human PET study, a clinically effective dose of telcagepant showed minimal receptor occupancy in the brain, suggestive for a lack of central action.52 Since earlier gepants with limited BBB permeability still achieved clinical efficacy, a peripheral site of action seems likely.53 TG seems to be a probable site of action for gepants but the actual target location, cell type, receptor type, and other central mechanism still need to be examined.
There were concerns that blocking CGRP’s vasodilatory effects could lead to vasoconstriction. In contrast to triptans that exhibit a risk of coronary vasoconstriction, gepants are a safer alternative. Ubrogepant and atogepant inhibited the vasodilatory responses to CGRP in the middle meningeal artery and coronary artery but exhibited no vasoconstrictive effect, with atogepant being a more potent vasodilatory antagonist than ubrogepant.54 An ex vivo study on rimegepant also showed no vasoconstriction of human coronary or cerebral arteries.55 However, although there is no vasoconstrictive effect, the long-term cardiovascular impact from chronic gepant use should still be investigated as long-term CGRP antagonism inhibits CGRP’s cardioprotective effect and may worsen cardiac dysfunction in chronic hypertensive mice.56
Acute Migraine Treatment
Ubrogepant has been studied for the treatment of acute migraine attacks (Table 1).
| Table 1. Summary of ubrogepant related clinical trials | ||
| Trial name | Dosage | Primary endpoint |
| NCT0161324857
Phase 2b, EM, n=527 |
100, 50, 25, 10, 1mg vs. placebo | 2-hr pain freedom (p<0.001 for trend test): 25.5%a, 21.0%b, 21.4%b, 14.8%, 5.6%, vs 8.9%. |
| NCT0282802058
Phase 3, EM, n=1327 |
100, 50mg vs. placebo | 2-hr pain freedom gain: OR 2.04 (95%CI 1.41-2.95), p<0.001; OR 1.83 (95%CI 1.25-2.66), p=0.002. 2-hr absence of MBS gain: OR 1.63 (95%CI 1.22-2.17), p=0.002; OR 1.70 (95%CI 1.27-2.28) p=0.002. |
| NCT0286770959
Phase 3, EM, n=1465 |
50, 25mg vs. placebo | 2-hr pain freedom gain: 7.5% (95%CI 2.6%-12.5%), p=0.01; 6.4% (95%CI 1.5%-11.5%), p=0.03. 2-hr absence of MBS gain: 11.5% (95%CI 5.4%-17.5%), p=0.01, 6.7% (95%CI 0.6%-12.7%), p=0.07 |
| ap<0.01 when compared to placebo, bp<0.05 when compared to placebo. EM: episodic migraine. MBS: most bothersome symptom. OR: odds ratio. | ||
In a phase IIb randomized, double-blind, placebo-controlled trial, 834 patients received either various doses of ubrogepant or placebo to treat one acute migraine attack. After two hours, there was a positive response trend in the proportion of patients achieving pain freedom (p<0.001), with ubrogepant 25mg, 50mg and 100 mg being significantly superior to placebo (21.4% vs. 8.9%, p=0.013; 21.0% vs. 8.9%, p=0.02; 25.5% vs. 8.9%, p=0.003).57 In addition, there have been two phase 3 trials (ACHIEVE I [NCT02828020] and II [NCT02867709]) that evaluated the efficacy, safety, and tolerability of ubrogepant compared to placebo in a single migraine attack in adults. In the ACHIEVE I trial, both ubrogepant 50mg and 100mg gained significantly more pain freedom at 2 hours (19.2%, 21.2%) than placebo (11.8%) and an increased absence of the most bothersome symptom at 2 hours (38,6%, 37.7%) than placebo (27.8%).58 In the ACHIEVE II trial, ubrogepant 25mg (20.7% vs. 14.3%, OR 1.56 [95%CI 1.09-2.22], p=0.03) and 50mg (21.8% vs. 14.3%, OR 1.62 [95%CI 1.14-2.29], p=0.01) both reported significant pain freedom at 2 hours. Therapeutic gains were 6.4% and 7.5% respectively. Absence of the most bothersome symptom and pain relief were also significantly different from placebo.59 In a post hoc analysis, ubrogepant 50mg was also responsive in triptan-ineffective subjects in regards to 2-hour pain freedom (16% vs. 8%, OR 2.16 [95%CI 1.19-3.95]) and absence of MBS (36% vs. 23%, OR 1.76 [95%CI 1.16-2.68]).60 In another post hoc analysis, the onset of pain relief for ubrogepant 50mg was achieved at 1 hour (43% vs. 37%, OR 1.3 [95%CI 1.06-1.59]).61 In an open-label 52-week extension trial, the therapeutic efficacy was maintained over the 1-year period.62 In a PK study, co-administration of ubrogepant and sumatriptan resulted in a 24% reduction of ubrogepant Cmax but the area under curve (AUC) remained unchanged.63 Co-administration of acetaminophen led to a 40% increase of ubrogepant Cmax and AUC but there was no change when co-administered with naproxen.64 These findings showed no clinically significant drug interaction with triptan and naproxen. In a study of high frequency dosing (2 days on and 2 days placebo alternating vs. all placebo), 7/516 subjects were found to have liver function test elevation ³ 3x the upper limit of normal (five placebo, two ubrogepant), with only two cases (one placebo, one ubrogepant) judged possibly related to treatment and one (ubrogepant) probably related. All were transient and resolved with continued dosing. Ubrogepant was well tolerated, and demonstrated a safety profile similar to placebo with the most common AEs being upper respiratory infection, nausea and dizziness but with no clinically relevant signs of hepatotoxicity.65 Ubrogepant is expected to receive an FDA decision in December this year.
Rimegepant has been studied for acute treatment of migraines (Table 2). In a phase 2 study comparing multiple dosages of rimegepant (BMS-927711 at that time) against placebo and sumatriptan, rimegepant at doses 75 mg, 150mg, 300mg, and 600mg but not 25mg and 10mg were found to have greater pain freedom at 2 hours when compared to placebo. Sumatriptan was more effective than rimegepant but the study was not designed for such comparison.66 In a multicenter, double-blind, phase 3 trial involving 1186 patients who were randomized to receive either rimegepant or placebo after a single migraine attack, significantly more subjects in the rimegepant group were pain-free 2 hours after treatment compared to placebo (19.6% vs. 12.0%, p<0.001); therapeutic gain was 7.6%. In addition, the percentage of patients free from their most bothersome symptom two hours after treatment was 37.6% in the rimegepant group vs. 25.2% in the placebo group (p<0.001); therapeutic gain was 12.4%.67 Interestingly, in 2 patients who started erenumab after 2 and 6 months of rimegepant usage, acute rimegepant still treated their breakthrough migraines successfully.68 The actual utility of gepants on top of CGRP mAbs remains to be examined and could be an open-label placebo effect. Another multicenter, double-blind, randomized phase 3 trial assigned 1466 patients to receive either 75 mg of rimegepant orally disintegrating tablet (ODT) or placebo to treat a single migraine attack of moderate or severe pain intensity. At 2 hours post-dose, rimegepant was superior to placebo in both freedom from pain (21% vs. 11%, therapeutic gain 10.4%, p <0.0001) and freedom from the most bothersome symptom (35% vs. 27%, therapeutic gain 8.3%, p=0.0009). Significant pain relief was also observed at post-dose 1 hour (p<0.05). The most common AEs were nausea and urinary tract infection, and no serious AE was reported.69 It is worth mentioning that there was a 2-fold increase in AUC and Cmax in subjects with severe hepatic impairment (Child-Pugh score 10-15) but no difference in subjects with mild to moderate hepatic impairment (Child-Pugh score 5-9) when compared to matched controls;70 dose adjustment in severe hepatic impaired patients is likely needed. In addition, vazegepant, which is administered intranasally using the Aptar Pharma Unidose System, has just completed its phase 2/3 trial (NCT03872453) but the result has not been published yet.
| Table 2. Summary of rimegepant related clinical trials | ||
| Trial name | Dosage | Primary endpoints |
| NCT0143044266
Phase 2b, EM, n=799 |
600, 300, 150, 75, 25, 10mg sumatriptan vs. placebo | 2-hr pain freedom: 20.7%a, 36.1%a, 28.2%a, 27.9%a, 16.4%, 12.7%, 26.0%a vs. 7.4%. |
| NCT0323784567
Phase 3, EM, n=1186 |
75mg vs. placebo | 2-hr pain freedom gain: 7.6% (95%CI 3.3%-11.9%)b
2-hr absence of MBS gain: 12.4% (95%CI 6.9%-17.9%)b |
| NCT0346175769
Phase 3, EM, n=1351 |
75mg vs. placebo | 2-hr pain freedom gain: 10.4% (95%CI 6.5%-14.2%)b
2-hr absence of MBS gain: 8.3% (95%CI 3.4%-13.2%)b |
| a p<0.01 when compared to placebo. b p<0.001 when compared to placebo | ||
Overall, these trials demonstrated clinical efficacy of gepants for acute migraine management with 5-10% of therapeutic gain when compared to placebo (Figure 1). Such gain seems slightly lower than those reported from acetaminophen or triptans.71 Gepants may benefit those with poor response to triptans.72 With their superior AE profile, gepants are an alternative to triptans. However, the clinically relevant benefit of gepants over triptans is yet to be seen once gepants become available. Whether gepants work acutely for patients with CM or in the presence of CGRP mAbs remains to be studied.
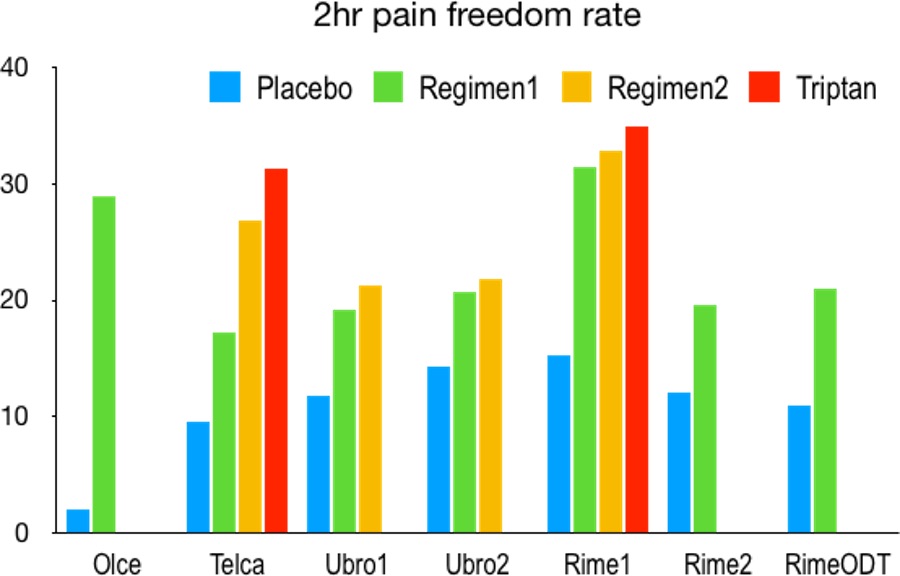
Migraine Prevention
Atogepant is an oral CGRP receptor antagonist in development for the prevention of migraine. It has a higher potency and longer half-life compared to ubrogepant. In a phase 2b/3 clinical trial, 795 patients with EM were randomized to placebo or various doses of atogepant and treated for 12 weeks under double-blind conditions for migraine prevention. All active treatment groups showed a statistically significant reduction from baseline in mean monthly migraine/probably migraine days (-0.7 to -1.39 days) across the 12-week treatment period. The most common AEs were nausea, fatigue, constipation, nasopharyngitis, and urinary tract infection. Liver safety profile (LFT >3x ULN) was similar to placebo (0.6-2.2% vs. 1.7%).73 In a post hoc analysis, therapeutic gain was 12-22% for ³50% responders rate (significant for 30mg QD, 30mg BID, 60mg BID).74
Rimegepant has also been studied for long-term use. In an interim analysis, 2867 subjects with 2-14 monthly migraine attacks were assigned to use rimegepant as needed (up to 1x/day) for up to 52 weeks with a subgroup getting scheduled every other day (QOD) dosing supplemented by as needed dosing for 12 weeks. Rimegepant was well tolerated with major treatment-associated AEs (³3%) being upper respiratory tract infection (8.5%), nasopharyngitis (6.4%), sinusitis (4.8%), urinary tract infection (3.8%), and influenzas (3.0%). 2.7% discontinued due to AEs. In 244 subjects who received the QOD regimen, 48.4% had a ³50% reduction in mean monthly migraine days (MMDs).75
Overall, gepants are a new orally available migraine preventive with minimal AEs. They can be an attractive alternative for patients who cannot tolerate conventional migraine preventives or who cannot receive CGRP mAbs. The therapeutic gain (-0.7 to -1.39 days) seems less than that from CGRP mAbs but the clinical dosage has not been optimized. It remains to be studied against other oral preventives or CGRP mAbs regarding clinical efficacy. Whether frequent use of gepants leads to medication overuse headache should be examined as well. At the time of writing, both rimegepant (NCT03732638) and atogepant (NCT02848326) are currently under investigation for migraine prevention.
CGRP-Targeting Monoclonal Antibodies
Monoclonal antibodies (mAbs) that target either the CGRP ligand or the CGRP receptor have been developed. Table 3 lists three CGRP-targeting mAbs that are FDA approved for the prevention of migraine (either chronic or episodic, with or without aura): erenumab, fremanezumab, and galcanezumab, along with eptinezumab, which is currently undergoing FDA review. All are humanized mAbs targeting the CGRP ligand except for erenumab, which is a fully human mAb targeting the CGRP receptor. They are indicated only for preventive use due to their slow absorption profile. Eptinezumab is the only one with IV formulation, and has a potential for acute treatment but has yet to be confirmed.
| Table 3. CGRP targeting monoclonal antibodies | |||||||
| Name | Sponsor | IgG | Route | T1/2 | Tmax | Dose | Frequency |
| Erenumab | Amgen | IgG2 | SC | 28D | 6D | 70mg
140mg |
QM
QM |
| Fremanezumab | Teva | IgG2 | SC | 32D | 5D | 225mg, 675mg | QM, QLT |
| Galcanezumab | Eli Lilly | IgG4 | SC | 27D | 5D | 120mga | QM |
| Eptinezumab | Lundbeck | IgG1 | IV | 26D | 1-3h | 100mg
300mg |
QLT
QLT |
| a Start with 240mg loading
IV: intravenous. SC: subcutaneous. QLT: quarterly. QM: monthly. |
|||||||
General Behavior Of Therapeutic Antibodies
An antibody is an immunoglobulin (Ig) produced by B cells. There are 5 subtypes, namely a (IgA), d (IgD), e (IgE), g (IgG), and m (IgM), with different structural properties. IgD and IgM act as antigen receptors on B cell membranes. IgG is a monomer, IgA is a dimer, and IgM is a pentamer. Among them, IgG constitutes 70% of all immunoglobulins in the body and is the most commonly used therapeutic antibody. It is made up of 2 light chains and 2 heavy chains joined by a disulfide bridge with a mass of 150 kDa (~7-10 nm). It is structurally divided into 3 parts (2 Fab and 1 Fc regions). The Fab parts contain complementarity-determining regions (CDRs) allowing for specific antigen binding that determines its major pharmacological function, and the Fc part plays a major role in activating effector cells and recycling antibodies from degradation. When the antibodies exhibit monovalent affinity, they are called mAbs. Using an immortalized hybridoma, a fusion of antibody-producing B cells and immortalized myeloma cells, Milstein and Köhler pioneered the production of mAbs in 1975 and later won the Nobel prize in physiology in 1984. However, the earlier therapeutic mAbs produced from murine antibodies unfortunately triggered a strong immune reaction in humans. Chimerization and humanization were developed to lower the foreign content thereby reducing the immunogenicity. Traditionally, chimeric antibodies (-ximab) are defined by having <85% human sequence on V gene, while humanized antibodies (-zumab) have ³85% derived from human DNA with only the CDR of the variable domains being foreign (e.g., non-human or synthetic). To further reduce the immunogenicity, fully human antibodies (-umab) were developed using technologies such as phage display and transgenic mice. However, many other antibody engineering techniques are employed as well, rendering the definition of “human” ambiguous. Thus, the WHO and American Medical Association proposed in 2017 a new naming system based not on the species but on the target, such as -ne- for neural or -ta- for tumor.76 Without the species information, the protein properties and immunogenic risks should therefore be documented separately.
The mechanism of action of therapeutic mAbs depends on what and where they bind, i.e., the pharmacodynamics (PD) and pharmacokinetics (PK). Since mAbs are structurally different from small molecules, their physical properties (e.g., size, charge, glycosylation pattern, target/off-target affinity) and chemical properties (e.g., degradation, metabolites, clearance) are strikingly distinct from small molecules. Similar to small molecules that bind to receptors or ion channels creating an agonistic or antagonistic effect, mAbs bind to ligands or receptors blocking their targets from action. Conversely, on immune cells, mAb binding enhances effector functions such as antibody-dependent cell-mediated cytotoxicity, antibody-dependent cellular phagocytosis, and complement-dependent cytotoxicity. To date, most mAbs are created with high target specificity in the picomolar range and basically no off-target affinity. The distribution of mAbs thus play a major role in the mechanism of action. Keep in mind there is a significant variation in PK between individuals even with the same mAbs. Here, we discuss some basic PK behavior of mAbs.77
Absorption
The route of administration determines the mechanism of absorption. Since protein gets degraded in the gastrointestinal tract, mAbs are typically administered IV or subcutaneously (SC). Only newborns can absorb antibody through GI absorption. Compared to IV injection, SC injection is more convenient but may be more painful and have lower bioavailability. The large size, positive surface charge, local proteolysis, and immunophagocytosis hinder the convection of mAbs to traverse SC tissue before entering the lymphatic system and circulation. Typically, it takes 3-7 days postdosing after SC injection to reach maximal serum concentration with 50-90% bioavailability.
Distribution
Once in the circulation, unlike small molecule that diffuse through blood vessel and tissue, mAbs extravasate and convect through vessel and tissue. Extravasation depends on the hydrostatic/oncotic pressure and the size of the paracellular pores in the vascular epithelium. This is a rather passive convective process, but sometimes receptor-mediated transcytosis may play a role. In a tight endothelium, the mAb tissue:blood ratio is usually low. In contrast, mAb leaks out in areas with fenestrated capillaries or sinusoid. This explains why mAbs are distributed mostly in organs such as the thyroid (67.5%), skin (15.7%), and liver (12.1%) but much less in the brain (0.35%).78 Once in the tissue, mAbs convect through the extracellular matrix (comprising of a negatively-charged scaffold that hinders the transport of positively charged mAbs) until they bind to the target, get metabolized, or recirculate back to the lymphatic system. In other words, mAbs that are retained in the tissue either bind to the targets or wait for the targets to appear before being eliminated; non-bound or non-trapped IgGs then convect back to the surrounding leaky veins or lymphatic ducts before entering the circulation. In the central nervous system, the dura mater, pituitary, circumventricular organs, and ganglia are much leakier than the blood-brain barrier or brain-CSF barrier (arachnoid mater). This explains why there is minimal accumulation of mAbs in the brain parenchyma or CSF. In contrast, sensory ganglia (TG, cervical, vagus, sphenopalatine, etc.) are highly permeable but interganglionic differences exist among them.79 Since all these ganglia play certain roles in the headache pathogenesis, they are hence presumably the action sites for CGRP mAbs used for headache. Keep in mind that even though the IgG accumulation in dura (11%) and TG (5.2%) are much higher than that in cortex (0.23%), hypothalamus (0.34%), or CSF (0.12%),80 IgG is not completely devoid in the parenchyma. Whether that small amount of IgG creates a centrally acting mechanism remains to be determined. Nonetheless, it is reasonable to believe that mAbs’ mechanism of action depends heavily on their distribution.
Elimination
IgG elimination usually takes place inside cells. It cannot be filtered by the kidneys nor metabolized by the liver. Rather, IgG gets taken up into cells by either receptor-mediated endocytosis (minor) or nonspecific pinocytosis (major). Receptor-mediated endocytosis takes place at the target cells’ surface (target-mediated drug disposition) or the immune cells’ surface that expresses Fc-gamma-receptors (FcgR). An example of this is the internalization of the antibody-receptor complex through membrane trafficking. These processes, however, play a minor role in IgG elimination. In contrast, pinocytosis is a nonspecific fluid-phase endocytosis utilized by endothelial cells throughout the body. It was estimated that skin, muscle, liver, and gut account for approximately 33, 24, 16, and 12% of IgG elimination;81 therefore, body surface area or body weight is often a common factor to determine mAb dosing. Since pinocytosis takes all the surrounding proteins, it requires a protective mechanism to salvage IgGs. This salvage pathway is mediated by neonatal Fc receptor (FcRn), which is also called the Brambell receptor. In neonates, FcRn mediates IgG transport from the mother via placental and intestine. FcRn is highly expressed in skin, muscle, liver, and spleen where vascular endothelia and reticuloendothelial cells are the major sites of IgG homeostasis.82 FcRn is also expressed in the brain microvascular endothelium and choroid plexus epithelium, facilitating the transport of IgG from the brain back to vessels.83 The mechanism of such a salvage pathway relies heavily on the IgG-FcRn affinity difference upon pH change. In the acidified endosome (i.e., low pH), FcRn binds to IgG. During exocytosis, IgG-FcRn returns to the cell surface and then releases the IgG at physiological pH. This recycling process is faster than IgG production and accounts for the relatively long half-life of the IgG, particularly IgG1, IgG2, and IgG4.84 Without FcRn, IgG catabolism is 10-fold faster.85 Higher IgG concentrations can also saturate the FcRn recycling pathway thereby increasing the IgG clearance rate. This explains why higher dosing with longer intervals may perform worse than lower dosing with shorter intervals. Besides intracellular degradation, immunogenicity to mAbs generates anti-drug antibodies (ADA), even if the mAb is completely analogous to human IgG. ADA is usually a polyclonal response that can be either neutralizing or non-neutralizing. Neutralizing ADAs eliminate the specific binding to the target; non-neutralizing ADAs may form ADA-mAb immune complexes that can increase clearance via the reticuloendothelial system.
With more understanding of the typical behavior of IgG, we can now discuss the mechanism of action of CGRP mAbs. Unfortunately, there are not many studies addressing this fundamental issue. CGRP mAbs likely act in a similar way to gepants by blocking CGRP’s function but exhibit certain unique features. While galcanezumab and erenumab bind to their targets reversibly, fremanezumab and eptinezumab engage the CGRP ligand irreversibily.86, 87 Additionally, in a cortical spreading depression rodent model, CGRP mAb was shown to selectively inhibit the responsiveness of Ad-fibers but not C-fibers of afferent meningeal nociceptors.88 CGRP mAb did not prevent CSD-induced arterial dilatation and plasma protein extravasation but prevented CGRP-induced arterial dilatation; thus, the CSD model likely differs from the CGRP infusion model.89 CGRP mAbs (either ligand or receptor) are believed to elicit no effect in the absence of CGRP. Rather, they interrupt CGRP-induced cAMP accumulation and, at a 10-fold higher dosage, receptor internalization.90 CGRP mAbs prevented the development of basal hyperalgesia and cutaneous mechanical hypersensitivity,91 and also inhibited bright light stress- and NO donor-induced cutaneous allodynia in animals previously primed with sumatriptan or morphine.92 These mAbs have been shown to inhibit neurogenic vasodilatation mediated by CGRP without affecting heart rate or arterial blood pressure in rats.93
Acute Migraine Treatment
Due to their slow absorption kinetic, there is no acute treatment trial involving the SC-injected CGRP function-blocking mAbs. In contrast, eptinezumab, which is administered IV with onset of action possibly within a few hours,91 is currently under investigation for subjects experiencing acute migraine attack (NCT04152083).
Migraine Prevention
Erenumab is a fully human mAb that targets the CGRP receptor. Erenumab was not associated with vasoactive properties per se but specifically inhibits CGRP-induced relaxation.94 It has been studied in the prevention of both CM and EM (Table 4). For CM, one phase 2, randomized, double-blind, multicenter study enrolled 667 patients with CM and randomly assigned them to either placebo, erenumab 70 mg, or erenumab 140 mg given every 4 weeks for 12 weeks. At the end of the study period, both erenumab 70 mg and 140 mg reduced MMDs compared to placebo (both doses -6.6 days vs. placebo -4.2 days, p<0.0001). The safety profile was similar to placebo, with the most frequent AEs being injection-site pain, upper respiratory tract infection, and nausea. 95 There are also several studies investigating erenumab’s efficacy in preventing EM. In one multicenter, randomized, double-blind phase 2 trial, 483 patients with EM were randomized to SC injections of placebo, erenumab 7 mg, erenumab 21 mg, or erenumab 70 mg in monthly doses for a total of 12 weeks. There was a significant mean change in MMDs at week 12 with erenumab 70 mg compared to placebo (-3.4 days vs -2.3 days, p=0.021).
| Table 4. Summary of erenumab related clinical trials | ||
| Trial name | Dosage | Primary endpoint |
| NCT0206641595
Phase 2, CM, n=667 |
140, 70mg vs. placebo | MMD reduction: both -2.5 (95%CI -3.5 to -1.4), p<0.0001 |
| NCT0195257496
Phase 2, EM, n=483 |
70, 21, 7mg vs. placebo | MMD reduction: -1.1 (95%CI -2.1 to -0.2), p=0.021; -0.1 (95%CI -1.1 to 0.9), p=0.83; 0.1 (95%CI -0.8 to 1.1), p=0.82 |
| NCT0248358597
Phase 3, EM, n=577 |
70mg vs. placebo | MMD reduction: -1.0 (95%CI -1.6 to -0.5), p<0.001. |
| NCT0245674098
Phase 3, EM, n=955 |
140, 70mg vs. placebo | MMD reduction (by 6 months): -1.9 (95%CI -2.3 to -1.4), p<0.001; -1.4 (95%Cl -1.9 to -0.9), p<0.001. |
| NCT0309683499
Phase 3b, EM*, n=246 |
140mg vs. placebo | ³50% responder rate: 30% vs. 14%, OR 2.7 (95%CI 1.4-5.2), p=0.002. |
| All studies were evaluated over 12-week unless otherwise specified. EM: episodic migraine. CM: chronic migraine. MMD: mean monthly migraine day. OR: odds ratio. * failed 2-4 migraine preventives. | ||
However, there was no significant difference between the 7 mg and 21 mg with placebo 96. In addition, a phase 3, randomized, double-blind study known as the ARISE trial was conducted in which 577 patients with EM were randomized to monthly SC injection of placebo or erenumab 70 mg for 12 weeks. At the end of the study period, those receiving erenumab experienced a mean -2.9 days change from baseline in MMDs compared to -1.8 days in placebo (p<0.001). Participants in the erenumab group also had an increased number of people who experienced a ³50% reduction in MMDs (40% vs. 30% placebo, p=0.010) and a greater reduction in monthly acute migraine-specific medication use (-1.2 days vs. -0.6 days placebo, p=0.002) 97. Another phase 3, randomized, double-blind study known as the STRIVE trial enrolled 955 patients with EM who were randomized to receive monthly SC injection of placebo or erenumab 70 mg or 140 mg for 24 weeks. There was a significant reduction in MMDs in both the 70 mg and 140 mg erenumab groups compared to placebo (-3.2 and -3.7 respectively vs. -1.8 placebo, p<0.0001 for both). In addition, a ³50% reduction in MMDs was achieved by 43% and 50% of the erenumab 70 mg and 140 mg groups compared to 27% in the placebo group (p<0.0001 for both) 98. For subjects who failed 2-4 preventive medications, at week 12, 36 (30%) patients in the erenumab had a 50% or greater reduction from baseline in the mean number of MMDs, compared with 17 (14%) in the placebo group. MMDs were reduced 1.6 days (95%CI -2.7 to -0.5; p=0.004). The tolerability and safety profiles of erenumab and placebo were similar. The most frequent treatment-emergent AE was injection site pain, which occurred in seven (6%) participants in both groups.99 At week 64 (52 weeks of open-label), 85% completed the week 52 visit. Overall, 47.1% achieved ³50% reduction in MMD and a MMD reduction of -3.7±4.1 days.100 In an interim analysis from an open-label study lasting 4+ years, MMDs were reduced by 5.8±3.8 days and acute medication days were reduced 4.6±3.3 days. The most common AEs were nasopharyngitis (10.9%), upper respiratory tract infection (6.8%), and influenza (4.7%) but there was no worsening of constipation.101, 102 However, serious constipation requiring hospitalizations was reported in post-marketing analysis.; “constipation with serious complications” was then added to the Aimovig FDA label.
Fremanezumab, a humanized mAb that targets both the a- and b-CGRP ligands, has been studied for the prevention of both CM and EM (Table 5). It has been shown to block CGRP-induced dilatation in human cerebral, middle meningeal, and abdominal arteries.103 In one multicenter, randomized, double-blind phase 2b study investigating fremanezumab for preventive treatment of CM, 264 patients were randomly assigned to receive either placebo or 2 different treatment regimens of fremanezumab SC for 3 28-day treatment cycles (either 675 mg in the first treatment cycle and 225 mg in the second and third cycles or 900 mg in all three cycles). Primary endpoints were change from baseline in number of headache-hours during the third treatment cycle (weeks 9-12) and safety and tolerability. Both treatment groups showed significant reduction in headache-hours compared to placebo, with a least square mean difference of -22.74 hours between the placebo and 675/225 mg group (p=0.0386) and -30.41 hours between the placebo and 900 mg group (p=0.0057). The most common AEs were mild injection-site pain and pruritis.104 A similar multicenter, randomized, double-blind phase 2b study also investigated the use of fremanezumab in preventing EM. 297 patients with high-frequency EM were enrolled and randomized to either placebo or 2 different doses of fremanezumab SC for 3 28-day treatment cycles (225 mg or 675 mg). Primary endpoints were change from baseline in migraine days during the third treatment cycle (weeks 9-12) and safety and tolerability. Results showed significant reductions in migraine-days from baseline between both treatment groups compared to placebo, with a least square mean change of -2.81 days between the 225 mg dose group and placebo (p<0.0001) and -2.64 days between the 675 mg dose and placebo (p<0.0001). AE incidence was similar between placebo and treatment groups.105
| Table 5. Summary of fremanezumab related clinical trials | ||
| Trial name | Dosage | Primary endpoint |
| NCT02021773104
Phase 2b, CM, n=264 |
900, 675/225mg vs. placebo | Headache hours: -30.41 (95%CI -51.88 to -8.95), P=0.0057; -22.74 (95%CI -44.28 to -1.21), p=0.0386 |
| NCT02025556105
Phase 2b, EM, n=297 |
675, 225mg, vs. placebo | MMD reduction: -2.64 (95%CI -3.9 to -1.38), p<0.0001; -2.81 (95%CI -4.07 to -1.55), p<0.001 |
| NCT02621931106
Phase 3, CM, n=376 |
675 once, 675/225mg, vs. placebo | MHD reduction: -1.8 (95%CI -2.4 to -1.2), p<0.001; -2.1 (95%CI -2.7 to -1.5), p<0.001 |
| NCT02629861107
Phase 3, EM, n=875 |
675 once, 675/225mg vs. placebo | MMD reduction: -1.3 (95%CI -1.79 to -0.72), p<0.001; -1.5 (95%CI -2.01 to -0.93), p<0.001 |
| NCT03308968109
Phase 3b, EM/CM*, n=838 |
675 once, 675/225mg vs. placebo | MMD reduction: -3.1 (95%CI -3.8 to -2.4), p<0.0001; -3.5 (95%CI -4.2 to -2.8), p<0.0001 |
| All studies were evaluated over 12 weeks. EM: episodic migraine. CM: chronic migraine. MMD: mean monthly migraine day. MHD: mean monthly headache day. * Failed 2-4 classes of migraine preventives | ||
In addition, fremanezumab has been studied in two 16-week, multicenter, randomized, double-blind phase 3 trials, known as the HALO studies, for the prevention of both CM and EM. In the CM study, 1130 patients were randomized to receive SC injections of either fremanezumab 675 mg at initiation followed by monthly 225 mg for 2 months (monthly dose regimen), fremanezumab 675 mg at initiation followed by placebo for two months (quarterly dose regimen), or three monthly doses of matching placebo. At the end of the study period, patients treated with fremanezumab experienced statistically significant reduction in mean monthly headache days (MHDs) of at least moderate severity compared to placebo (-2.5 days) during the 12-week period after the first dose for both monthly (-4.6 days, p<0,0001) and quarterly (-4.3 days, p<0.0001) dosing regimens. The most common AE was injection site pain, with similar rates in the placebo and active groups.106 In the EM study, 875 patients were randomized to receive SC injections of fremanezumab 225 mg as a monthly dose for 3 months, 675 mg at initiation followed by placebo for 2 months, or 3 monthly doses of matching placebo. At the end of the study period, fremanezumab given monthly resulted in a 41.6% reduction in migraines relative to baseline (-3.7 days vs. -2.2 days for placebo, p<0.0001) and the quarterly dose of fremanezumab also showed significant improvement in migraine days (-3.4 days or 37.0%, p<0.0001).107 In a post hoc analysis of both HALO studies, it was shown that the effect was observed at week 1 (-0.5 days [95%CI -0.7 to -0.3], p<0.0001).108 In a recent study involving 838 subjects (EM 39%, CM 61%) who failed 2-4 classes of migraine preventives with similar treatment assignment as the HALO study, reductions from baseline in monthly average migraine days over 12 weeks were greater versus placebo (least-squares mean [LSM] change −0.6 [SE 0.3]) with quarterly fremanezumab (LSM change −3.7 [0.3]; LSM difference vs placebo −3.1 [95% CI −3.8 to −2.4]; p<0.0001) and with monthly fremanezumab (LSM change −4.1 [0.34]; LSM difference vs placebo −3.5 [−4.2 to −2.8]; p<0.0001). AEs were similar for placebo and fremanezumab. Serious AEs were reported in four (1%) of 277 participants with placebo, two (<1%) of 276 with quarterly fremanezumab, and four (1%) of 285 with monthly fremanezumab.109 Interestingly, the therapeutic gains were much higher than that from the HALO studies, most likely due to low placebo response.
Galcanezumab is another humanized mAb antagonist that targets the a- and b-CGRP ligands (Table 6). In a randomized, multicenter, double-blind, phase 2 proof-of-concept study, 218 patients with migraine were randomly assigned to a SC injection of either galcanezumab 150 mg or placebo every two weeks for twelve weeks. At the end of twelve weeks, there was a significant reduction from baseline in the number of migraine headache days in the galcanezumab group compared to placebo (-4.2 days vs. -3.0 days, least-squared mean difference -1.2, p=0.0030). AEs that occurred more frequently with galcanezumab included injection site pain, erythema, upper respiratory infections, and abdominal pain. No serious AEs were found that were related to the study drug. 110 Two phase 3, randomized, double-blind, controlled clinical trials titled EVOLVE-1 and EVOLVE-2 were also conducted to investigate the efficacy and safety of galcanezumab for the prevention of EM. In EVOLVE-1, 858 patients with EM were randomized (2:1:1) to monthly SC injection of placebo, galcanezumab 120 mg, or galcanezumab 240 mg for 6 months. Treatment with galcanezumab significantly reduced MMDs in both the galcanezumab 120 mg and 240 mg groups compared to placebo (-4.7 days and -4.6 days respectively vs. -2.8 days placebo, p<0.001 for both). The incidence of discontinuation due to Aes was less than 5% across all treatment groups.111 In EVOLVE-2, 915 patients with EM were enrolled with the same treatment regimen as the EVOLVE-1 trial. After 6 months, MMDs were reduced by 4.3 and 4.2 days by galcanezumab 120 mg and 240 mg respectively compared to 2.3 days by placebo (p<0.001 for both). In addition, significantly greater mean proportions of patients in the galcanezumab 120 mg and 240 mg groups experienced ³50% reduction in migraine headache days compared to placebo (59% and 57% vs. 36%, p<0.001). Injection site pain was the most common AE and was reported at similar rates in all treatment groups, but both galcanezumab doses had significantly more injection site reactions and pruritis compared to placebo.112 In a post hoc study pooling EVOLVE 1 and 2 studies for those who failed ³2 prior preventives, overall in 6 months there were reductions of -2.60 days (95%CI-3.95 to -1.25) and -3.37 days (95%CI -4.78 to -1.96) for the 120mg and 240mg doses, respectively.113 Another phase 3 trial known as the REGAIN study evaluated the safety and efficacy of galcanezumab in the prevention of CM. In this study, 1113 patients with CM were randomized (2:1:1) to monthly SC injections of placebo, galcanezumab 120 mg (with a 240 mg loading dose), or galcanezumab 240 mg for a total of 3 months. After 3 months, both the galcanezumab 120 mg and 240 mg groups demonstrated greater overall mean reduction in the number of MMDs compared to placebo (-4.8 days and -4.6 days respectively vs. -2.7 days placebo, p<0.001 for both). There were no significant differences between the groups on any safety or tolerability outcomes other than a higher incidence of injection-site reaction, erythema, and pruritis as well as sinusitis in the galcanezumab 240 mg group relative to placebo 114. In a long-term open-label study (n=270), 77.8% completed the open-label phase, and 4.8% discontinued due to AEs. Overall the migraine days were reduced (-5.6 for 120mg, -.6.5 for 240mg). Treatment emergent AEs with a frequency ≥ 10% of patients in either dose group were injection site pain, nasopharyngitis, upper respiratory tract infection, injection site reaction, back pain, and sinusitis.115
| Table 6. Summary of galcanezumab related clinical trials | ||
| Trial name | Dosage | Primary endpoint |
| NCT01625988110
Phase 2, EM, n=218 |
150mg vs. placebo | MMD reduction: -1.2 (90%CI -1.9 to -0.6), p=0.003 |
| NCT02614183111
Phase 3, EM, n=858 |
240, 120mg vs. placebo | MMD reduction (at 6 month): -1.8 (95%CI -2.5 to -1.4), p<0.001; -1.9 (95%CI -2.5 to -1.4), p<0.001 |
| NCT02614196112
Phase 3, EM, n=915 |
240, 120mg vs. placebo | MMD reduction (at 6 month): -1.9 (95%CI -2.4 to -1.4), p<0.001; -2.0 (95%CI -2.6 to -1.5), p<0.001 |
| NCT02614261114
Phase 3, CM, n=1113 |
240, 120mg vs. placebo | MMD reduction: -1.9 (95%CI -2.7 to -1.1), p<0.001; -2.1 (95%CI -2.9 to -1.3), p<0.001 |
| All studies were evaluated over 12-week unless otherwise specified. EM: episodic migraine. CM: chronic migraine. MMD: mean monthly migraine day. | ||
Eptinezumab is a humanized mAb targeting both the a- and b-CGRP ligands and is produced by yeast as opposed to mammalian cells like the other mAbs. It has been studied for prevention of both CM and EM (Table 7). In a randomized, double-blind, placebo-controlled, exploratory, proof-of-concept phase 2 trial, 174 patients with EM were randomly assigned to receive either in intravenous dose of 1000 mg of eptinezumab or placebo and assessed for safety 12 weeks after infusion and change in baseline to weeks 5-8 in the frequency of migraine days. The mean change in migraine days between baseline and weeks 5-8 was statistically significant, with -5.6 days in the treatment group compared to -4.6 days in the placebo group (p=0.0306). The most common AEs included upper respiratory tract infection (URI; 9 vs. 7%), urinary tract infection (UTI; 1 vs. 5%), fatigue (4% both groups), back pain (4 vs. 5%), nausea and vomiting (4 vs. 2%), and arthralgia (1 vs. 5%) in the eptinezumab versus placebo groups, respectively.116 Another phase 2b, parallel-group, double-blind, randomized clinical trial investigated the safety, tolerability, and effectiveness of eptinezumab for the prevention of CM. 616 patients with CM were enrolled and randomized (1:1:1:1:1) to eptinezumab 10 mg, 30 mg, 100 mg, or 300 mg or placebo, administered as a single IV infusion with the primary endpoint being the percentage of patients with a ³75% decrease in MMDs over weeks 1-12. Patients in the eptinezumab 300 mg group had significant ³75% migraine responder rates compared to placebo (33.3% vs. 20.7%, p=0.033). However, patients in the eptinezumab 10 mg, 30 mg, and 100 mg did not achieve statistically significant results compared to placebo. AE rates were similar to placebo 117. Two randomized, double-blinded phase 3 trials also investigated the safety and efficacy of eptinezumab for the treatment of EM (PROMISE-I) and CM (PROMISE II). In PROMISE-I, 888 patients were randomized to eptinezumab 30 mg, 100 mg, 300 mg, or placebo by IV infusion once with the primary end point being the mean change from baseline in MMDs over the 12-week treatment period. Patients in the eptinezumab 100 mg, and 300 mg groups all had significant reductions in MMDs compared to placebo (-3.9 [p=0.0179], and -4.3 [p=0.0001] vs. -3.2, respectively). AEs for eptinezumab were similar to placebo 118. In PROMISE-II, 1072 patients with CM were randomized to eptinezumab 100 mg, 300 mg, or placebo administered intravenously every 12 weeks for 2 infusions with the primary endpoint being the change from baseline in MMDs over weeks 1-12. Patients in both eptinezumab 100 mg and 300 mg treatment groups showed significant reduction from baseline in MMDs compared to placebo over months 1-3 (-7.7 days and -8.2 days respectively vs. -5.6 days, p<0.0001 for both) and months 4-6 (-8.1 days and -8.8 days respectively vs. -6.1 days, p<0.0001 for both). Rates of AEs were again similar between groups.119 In the PREVAIL study, which is an open-label 1-year safety study, treatment emergent AEs were reported for 64.8% of patients, with the most common (≥5%) being nasopharyngitis (13.3%), upper respiratory tract infection (7.0%), sinusitis (6.3%), and influenza (5.5%).120
| Table 7. Summary of eptinezumab related clinical trials | ||
| Dosage | Primary endpoint | |
| NCT01772524116
Phase 2, EM, n=163 |
1000mg vs. placebo | MMD reduction: -1.0 (95%CI -2.0 to 0.1) |
| NCT02275117117
Phase 2b, CM, n=616 |
300, 100, 30, 10mg vs. placebo | ³75% responder rates: 33.3%, 31.4%, 28.2%, 26.8%, vs. 20.7% (p=0.033, 0.072, 0.201, 0.294). |
| NCT02559895118
Phase 3, EM, n=888 |
300, 100, 30mg vs. placebo | MMD changea: -4.3, -3.9, vs. -3.2 (p=0.0001, 0.0179) |
| NCT02974153119
Phase 3, CM, n=1072 |
300, 100mg vs. placebo | MMD change: -8.2, -7.7, vs. -5.6 (p<0.0001, <0.0001) |
| All studies were evaluated over 12-week unless otherwise specified. EM: episodic migraine. CM: chronic migraine. MMD: mean monthly migraine days. a Data from 30mg was not reported. | ||
Clinical Perspective
These clinical trials have demonstrated the clinical efficacy of CGRP mAbs. Keep in mind that these trials were conducted under different study populations and calculated differently for their primary endpoints; this inconsistency renders their comparison based on published trials impractical. Figure 2 illustrates the MMD changes reported in each study. Overall, CGRP mAbs reduced MMDs in subjects with EM (therapeutic gain 0.7-1.9 days) and CM (therapeutic gain 1.5-2.4 days). Acute medication use was also reduced in subjects with EM (therapeutic gain 0.5-3 days) and CM (therapeutic gain 1.9-2.5 days). In addition, less patients reported medication overuse (therapeutic gain 9-15%) after 12 weeks.121 Interestingly, subjects who previously failed preventive therapy exhibited lower placebo response and a higher therapeutic gain of 1.6-3.5 days.99, 109,113 The time to onset of action for IV- and SC-administered CGRP mAb are likely within 1 day and 1 week, respectively.118, 119,122, 123, 124 Early non-responders may still respond by later months.123 With a complementary mechanism and minimal drug interactions, CGRP mAbs likely will gain additional clinical benefit when used in combination with current preventive medications.125 The low number needed to treat (NTT) and high number needed to harm (NNH) of CGRP mAbs demonstrates a favorable benefit-risk profile against other migraine preventive therapies126 .
![]()
However, some practical issues remain.
- CGRP is a key neuropeptide in tissue healing, hematopoiesis, and the neuro-immune axis. Prolonged CGRP blockade may impact certain restorative functions, unmask underlying autoimmunity, or perhaps even worsen active infection or cardiac dysfunction.
- All trials included only patients in a relatively healthy state (BMI<40, age <70 years, no active major cardiovascular or other major health issues, not pregnant or breastfeeding). The clinical response in these unstudied yet vulnerable populations remain to be seen.
- IgG, particularly IgG1 and IgG4, is readily transported via FcRn across placenta and breast alveoli. CGRP mAb is probably unsafe for fetuses as CGRP plays a role in placental vascular adaptation and decidualization.127 Per FDA label, no AEs on offspring were observed when pregnant monkeys were administered erenumab throughout gestation. At this moment we do not have sufficient clinical data to justify its safety for use in women who are pregnant or breastfeeding.
- It has been our personal experience that a second mAb agent may be successful if the first one fails or has significant AEs.
- Insurance coverage and drug affordability remain important issues. Insurers typically require a failure of 2 classes of preventive medications or onabotulinum toxin (Botox) in order to approve coverage. Practically, a specialty pharmacy can be employed to reduce the financial and logistical burden to patients. However, combination therapy of CGRP mAbs with Botox or gepants is still not covered at this moment but hopefully will be in the future.
Conclusion
CGRP plays a fundamental role in migraine pathogenesis. CGRP antagonism, via either gepants or mAbs, has demonstrated clinical efficacy in both acute and preventive treatment of migraine. This is a new class of medication that for the first time ever was developed to be specifically tailored to the underlying pathophysiology of migraine. Gepants likely exhibit slightly less efficacy than triptans but have much less vasoconstrictive risk; long-term safety on their frequent use remains to be examined. CGRP and CGRP receptor mAbs significantly reduce MMDs, decrease acute medication use, and improve patient’s quality of life. They are effective even in those who failed multiple preventive medications. Their convenient monthly dosing and minimal AE profile not only improve medication compliance but may also reduce migraine chronification. However, keep in mind that they do not work for everyone as CGRP is not the only player in the pain pathways of migraine and its functional blockade may cause harm in certain vulnerable populations. More well-designed clinical trials are still needed.
Acknowledgement
None
Disclosures
Conflicts of interest: In compliance with the ICMJE uniform disclosure form, all authors declare the following: Dr. Yuan receives honoraria from Supernus Pharmaceuticals, Inc., Dr. Silberstein receives honoraria from Alder Biopharmaceuticals; Allergan, Inc.; Amgen; Avanir Pharmaceuticals, Inc.; Depomed; Dr. Reddy’s Laboratories; eNeura Inc.; electroCore Medical, LLC; Ipsen Biopharmaceuticals; Medscape, LLC; Medtronic, Inc.; Mitsubishi Tanabe Pharma America, Inc.; NINDS; St. Jude Medical; Supernus Pharmaceuticals, Inc.; Teva Pharmaceuticals and Trigemina, Inc.
References
- GBD 2017 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. 2018;392(10159):1789-1858. Pubmed CrossRef
- Hawkins K, Wang S, Rupnow M. Direct cost burden among insured US employees with migraine. 2008;48(4):553-563. Pubmed CrossRef
- Maniyar FH, Sprenger T, Monteith T, Schankin C, Goadsby PJ. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. 2014;137(Pt 1):232-241. Pubmed CrossRef
- Giffin NJ, Lipton RB, Silberstein SD, Olesen J, Goadsby PJ. The migraine postdrome: An electronic diary study. Neurology. 2016;87(3):309-313. Pubmed CrossRef
- Headache Classification Committee of the International Headache Society (IHS) The International Classification of Headache Disorders, 3rd edition. 2018;38(1):1-211. Pubmed CrossRef
- Probyn K, Bowers H, Caldwell F, et al. Prognostic factors for chronic headache: A systematic review. 2017;89(3):291-301. Pubmed CrossRef
- Hepp Z, Dodick DW, Varon SF, et al. Persistence and switching patterns of oral migraine prophylactic medications among patients with chronic migraine: A retrospective claims analysis. Cephalalgia. 2017;37(5):470-485. Pubmed CrossRef
- Edvinsson L. The Trigeminovascular Pathway: Role of CGRP and CGRP Receptors in Migraine. 2017;57 Suppl 2:47-55. Pubmed CrossRef
- Goadsby PJ, Holland PR, Martins-Oliveira M, Hoffmann J, Schankin C, Akerman S. Pathophysiology of Migraine: A Disorder of Sensory Processing. Physiol Rev. 2017;97(2):553-622. Pubmed CrossRef
- Eftekhari S, Salvatore CA, Calamari A, Kane SA, Tajti J, Edvinsson L. Differential distribution of calcitonin gene-related peptide and its receptor components in the human trigeminal ganglion. 2010;169(2):683-696. Pubmed CrossRef
- Knyihár-Csillik E, Tajti J, Chadaide Z, Csillik B, Vécsei L. Functional immunohistochemistry of neuropeptides and nitric oxide synthase in the nerve fibers of the supratentorial dura mater in an experimental migraine model. Microsc Res Tech. 2001;53(3):193-211. Pubmed CrossRef
- Buck SH, Walsh JH, Yamamura HI, Burks TF. Neuropeptides in sensory neurons. Life Sci. 1982;30(22):1857-1866. Pubmed CrossRef
- Russo AF. Overview of Neuropeptides: Awakening the Senses? 2017;57 Suppl 2:37-46. Pubmed CrossRef
- van den Pol AN. Neuropeptide transmission in brain circuits. 2012;76(1):98-115. Pubmed CrossRef
- Russell FA, King R, Smillie SJ, Kodji X, Brain SD. Calcitonin gene-related peptide: physiology and pathophysiology. Physiol Rev. 2014;94(4):1099-1142. Pubmed CrossRef
- Edvinsson L. The Journey to Establish CGRP as a Migraine Target: A Retrospective View. 2015;55(9):1249-1255. Pubmed CrossRef
- Hendrikse ER, Bower RL, Hay DL, Walker CS. Molecular studies of CGRP and the CGRP family of peptides in the central nervous system. 2019;39(3):403-419. Pubmed CrossRef
- Mulderry PK, Ghatei MA, Spokes RA, et al. Differential expression of alpha-CGRP and beta-CGRP by primary sensory neurons and enteric autonomic neurons of the rat. 1988;25(1):195-205. Pubmed CrossRef
- Messlinger K. The big CGRP flood – sources, sinks and signalling sites in the trigeminovascular system. J Headache Pain. 2018;19(1):22. Pubmed CrossRef
- Brain SD, Williams TJ, Tippins JR, Morris HR, MacIntyre I. Calcitonin gene-related peptide is a potent vasodilator. 1985;313(5997):54-56. Pubmed CrossRef
- Kraenzlin ME, Ch’ng JL, Mulderry PK, Ghatei MA, Bloom SR. Infusion of a novel peptide, calcitonin gene-related peptide (CGRP) in man. Pharmacokinetics and effects on gastric acid secretion and on gastrointestinal hormones. Regul Pept. 1985;10(2-3):189-197. Pubmed CrossRef
- McLatchie LM, Fraser NJ, Main MJ, et al. RAMPs regulate the transport and ligand specificity of the calcitonin-receptor-like receptor. 1998;393(6683):333-339. Pubmed CrossRef
- Yuan H, Spare NM, Silberstein SD. Targeting CGRP for the Prevention of Migraine and Cluster Headache: A Narrative Review. 2019;59 Suppl 2:20-32. Pubmed CrossRef
- Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28(2):183-187. Pubmed CrossRef
- Sarchielli P, Alberti A, Codini M, Floridi A, Gallai V. Nitric oxide metabolites, prostaglandins and trigeminal vasoactive peptides in internal jugular vein blood during spontaneous migraine attacks. 2000;20(10):907-918. Pubmed CrossRef
- Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33(1):48-56. Pubmed CrossRef
- Cernuda-Morollon E, Ramon C, Martinez-Camblor P, Serrano-Pertierra E, Larrosa D, Pascual J. OnabotulinumtoxinA decreases interictal CGRP plasma levels in patients with chronic migraine. 2015;156(5):820-824. Pubmed CrossRef
- Akerman S, Goadsby PJ. Topiramate inhibits trigeminovascular activation: an intravital microscopy study. Br J Pharmacol. 2005;146(1):7-14. Pubmed CrossRef
- Durham PL, Cady R, Cady R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: implications for migraine therapy. 2004;44(1):35-42; discussion 42-3. Pubmed CrossRef
- Hou M, Kanje M, Longmore J, Tajti J, Uddman R, Edvinsson L. 5-HT(1B) and 5-HT(1D) receptors in the human trigeminal ganglion: co-localization with calcitonin gene-related peptide, substance P and nitric oxide synthase. Brain Res. 2001;909(1-2):112-120. Pubmed CrossRef
- Classey JD, Bartsch T, Goadsby PJ. Distribution of 5-HT(1B), 5-HT(1D) and 5-HT(1F) receptor expression in rat trigeminal and dorsal root ganglia neurons: relevance to the selective anti-migraine effect of triptans. Brain Res. 2010;1361:76-85. Pubmed CrossRef
- Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. 2002;22(1):54-61. Pubmed CrossRef
- Hansen JM, Hauge AW, Olesen J, Ashina M. Calcitonin gene-related peptide triggers migraine-like attacks in patients with migraine with aura. 2010;30(10):1179-1186. Pubmed CrossRef
- Hansen JM, Thomsen LL, Olesen J, Ashina M. Calcitonin gene-related peptide does not cause migraine attacks in patients with familial hemiplegic migraine. 2011;51(4):544-553. Pubmed CrossRef
- Ashina M, Bendtsen L, Jensen R, Schifter S, Olesen J. Calcitonin gene-related peptide levels during nitric oxide-induced headache in patients with chronic tension-type headache. Eur J Neurol. 2001;8(2):173-178. Pubmed CrossRef
- Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;58(5):698-705. Pubmed CrossRef
- Russo AF. Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55(1):533-552. Pubmed CrossRef
- Afroz S, Arakaki R, Iwasa T, et al. CGRP Induces Differential Regulation of Cytokines from Satellite Glial Cells in Trigeminal Ganglia and Orofacial Nociception. Int J Mol Sci. 2019;20(3). Pubmed CrossRef
- Thalakoti S, Patil VV, Damodaram S, et al. Neuron-glia signaling in trigeminal ganglion: implications for migraine pathology. 2007;47(7):1008-1023; discussion 1024-1005. Pubmed CrossRef
- Capuano A, De Corato A, Lisi L, Tringali G, Navarra P, Dello Russo C. Proinflammatory-activated trigeminal satellite cells promote neuronal sensitization: relevance for migraine pathology. Mol Pain. 2009;5:43. Pubmed CrossRef
- De Corato A, Lisi L, Capuano A, et al. Trigeminal satellite cells express functional calcitonin gene-related peptide receptors, whose activation enhances interleukin-1beta pro-inflammatory effects. J Neuroimmunol. 2011;237(1-2):39-46. Pubmed CrossRef
- De Felice M, Ossipov MH, Wang R, et al. Triptan-induced latent sensitization: a possible basis for medication overuse headache. Ann Neurol. 2010;67(3):325-337. Pubmed CrossRef
- Olesen J, Diener HC, Husstedt IW, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350(11):1104-1110. Pubmed CrossRef
- Ho TW, Ferrari MD, Dodick DW, et al. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. 2008;372(9656):2115-2123. Pubmed CrossRef
- Ho TW, Mannix LK, Fan X, et al. Randomized controlled trial of an oral CGRP receptor antagonist, MK-0974, in acute treatment of migraine. 2008;70(16):1304-1312. Pubmed CrossRef
- Hewitt DJ, Aurora SK, Dodick DW, et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. 2011;31(6):712-722. Pubmed CrossRef
- Ho TW, Connor KM, Zhang Y, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. 2014;83(11):958-966. Pubmed CrossRef
- Filiz A, Tepe N, Eftekhari S, et al. CGRP receptor antagonist MK-8825 attenuates cortical spreading depression induced pain behavior. 2019;39(3):354-365. Pubmed CrossRef
- Sixt ML, Messlinger K, Fischer MJ. Calcitonin gene-related peptide receptor antagonist olcegepant acts in the spinal trigeminal nucleus. 2009;132(Pt 11):3134-3141. Pubmed CrossRef
- Walker CS, Raddant AC, Woolley MJ, Russo AF, Hay DL. CGRP receptor antagonist activity of olcegepant depends on the signalling pathway measured. 2018;38(3):437-451. Pubmed CrossRef
- Zhang Z, Winborn CS, Marquez de Prado B, Russo AF. Sensitization of calcitonin gene-related peptide receptors by receptor activity-modifying protein-1 in the trigeminal ganglion. J Neurosci. 2007;27(10):2693-2703. Pubmed CrossRef
- Hostetler ED, Joshi AD, Sanabria-Bohorquez S, et al. In vivo quantification of calcitonin gene-related peptide receptor occupancy by telcagepant in rhesus monkey and human brain using the positron emission tomography tracer [11C]MK-4232. J Pharmacol Exp Ther. 2013;347(2):478-486. Pubmed CrossRef
- Tfelt-Hansen P, Olesen J. Possible site of action of CGRP antagonists in migraine. 2011;31(6):748-750. Pubmed CrossRef
- Rubio-Beltran E, Chan KY, Danser AJ, MaassenVanDenBrink A, Edvinsson L. Characterisation of the calcitonin gene-related peptide receptor antagonists ubrogepant and atogepant in human isolated coronary, cerebral and middle meningeal arteries. 2019; doi:10.1177/0333102419884943. Pubmed CrossRef
- Conway CM, Croop R, Dubowchik GM, Coric V, Lipton RB. Cardiovascular Safety of Rimegepant 75 mg in 3 Randomized Clinical Trials and Systematic Evaluations from In Vitro, Ex Vivo, and In Vivo Nonclinical Assays. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Skaria T, Mitchell KJ, Vogel O, Walchli T, Gassmann M, Vogel J. Blood Pressure Normalization-Independent Cardioprotective Effects of Endogenous, Physical Activity-Induced alphaCGRP (alpha Calcitonin Gene-Related Peptide) in Chronically Hypertensive Mice. Circ Res. 2019;125(12):1124-1140. Pubmed CrossRef
- Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double-blind, placebo-controlled trial of ubrogepant for the acute treatment of migraine. 2016;36(9):887-898. Pubmed CrossRef
- Dodick DW, Lipton RB, Ailani J, et al. Ubrogepant for the Treatment of Migraine. N Engl J Med. 2019;381(23):2230-2241. Pubmed CrossRef
- Lipton RB, Dodick DW, Ailani J, et al. Effect of Ubrogepant vs Placebo on Pain and the Most Bothersome Associated Symptom in the Acute Treatment of Migraine: The ACHIEVE II Randomized Clinical Trial. 2019;322(19):1887-1898. Pubmed CrossRef
- Blumenfeld AM, Goadsby PJ, Dodick DW, et al. Ubrogepant Is Effective for the Acute Treatment of Migraine in Patients for Whom Triptans Are Ineffective. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Dodick DW, Goadsby PJ, Lakkis H, et al. Ubrogepant Achieves Onset of Pain Relief at 1 Hour for the Acute Treatment of Migraine. 61st Annual Scientific Meeting American Headache Society®; July 11-14, 2019; Philadelphia.
- Lipton RB, Ailani J, Hutchinson S, et al. Efficacy Is Maintained with Long-term Intermittent Use of Ubrogepant for the Acute Treatment of Migraine. 61st Annual Scientific Meeting American Headache Society®; July 11-14, 2019; Philadelphia.
- Jakate A, Boinpally R, Butler M, Lu K, McGeeney D, Periclou A. Coadministration of Single Therapeutic Oral Doses of Ubrogepant and Sumatriptan Produces No Clinically Relevant Pharmacokinetic Interactions. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Jakate A, Boinpally R, Butler M, et al. Single Therapeutic Doses of Ubrogepant Are Not Associated with a Clinically Relevant Drug-Drug Interaction When Co-administered with Acetaminophen or Naproxen. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Goadsby PJ, Tepper SJ, Watkins PB, et al. Safety and tolerability of ubrogepant following intermittent, high-frequency dosing: Randomized, placebo-controlled trial in healthy adults. 2019;39(14):1753-1761. Pubmed CrossRef
- Marcus R, Goadsby PJ, Dodick D, Stock D, Manos G, Fischer TZ. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. 2014;34(2):114-125. Pubmed CrossRef
- Lipton RB, Croop R, Stock EG, et al. Rimegepant, an Oral Calcitonin Gene-Related Peptide Receptor Antagonist, for Migraine. N Engl J Med. 2019;381(2):142-149. Pubmed CrossRef
- Mullin K, Kudrow D, Croop R, et al. Acute Treatment Benefit from Oral CGRP Receptor Antagonist and Monoclonal Antibody Combination: Rimegepant 75 mg for Acute Treatment of Attacks During Preventive Therapy With Erenumab. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Croop R, Goadsby PJ, Stock DA, et al. Efficacy, safety, and tolerability of rimegepant orally disintegrating tablet for the acute treatment of migraine: a randomised, phase 3, double-blind, placebo-controlled trial. 2019;394(10200):737-745. Pubmed CrossRef
- Croop R, Stringfellow J, Ivans A, Coric V. Rimegepant 75 mg in Subjects with Hepatic Impairment: Results of a Phase 1, Open-label, Single-dose, Parallel-group Study. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Tfelt-Hansen P, Loder E. The Emperor’s New Gepants: Are the Effects of the New Oral CGRP Antagonists Clinically Meaningful? 2019;59(1):113-117. Pubmed CrossRef
- Ho TW, Olesen J, Dodick DW, Kost J, Lines C, Ferrari MD. Antimigraine efficacy of telcagepant based on patient’s historical triptan response. 2011;51(1):64-72. Pubmed CrossRef
- Goadsby PJ, Dodick DW, Ailani J, et al. Orally Administered Atogepant Was Efficacious, Safe, and Tolerable for the Prevention of Migraine: Results from a Phase 2b/3 Study. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Dodick DW, Ailani J, Goadsby PJ, et al. Responder Rates to Atogepant in Patients with Episodic Migraine: A Post Hoc Analysis of Results from a Phase 2b/3, Randomized, Double-Blind, Placebo-Controlled Trial. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Phiadelphia.
- Lipton RB, Berman G, Kudrow D, et al. Long-Term, Open-Label Safety Study of Rimegepant 75 mg for the Treatment of Migraine (Study 201): Interim Analysis of Safety and Exploratory Efficacy. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Mayrhofer P, Kunert R. Nomenclature of humanized mAbs: Early concepts, current challenges and future perspectives. Hum Antibodies. 2019;27(1):37-51. Pubmed CrossRef
- Ryman JT, Meibohm B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst Pharmacol. 2017;6(9):576-588. Pubmed CrossRef
- Shah DK, Betts AM. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. 2013;5(2):297-305. Pubmed CrossRef
- Anzil AP, Blinzinger K, Herrlinger H. Fenestrated blood capillaries in rat cranio-spinal sensory ganglia. Cell Tissue Res. 1976;167(4):563-567. Pubmed CrossRef
- Johnson KW, Morin SM, Wroblewski VJ, Johnson MP. Peripheral and central nervous system distribution of the CGRP neutralizing antibody [(125)I] galcanezumab in male rats. 2019;39(10):1241-1248. Pubmed CrossRef
- Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinet Pharmacodyn. 2007;34(5):687-709. Pubmed CrossRef
- Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179(7):4580-4588. Pubmed CrossRef
- Schlachetzki F, Zhu C, Pardridge WM. Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J Neurochem. 2002;81(1):203-206. Pubmed CrossRef
- Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol. 2011;22(6):868-876. Pubmed CrossRef
- Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal transport receptor. Proc Natl Acad Sci U S A. 1996;93(11):5512-5516. Pubmed CrossRef
- Karasek C OE, Allison D, Latham J. Characterization of the intrinsic binding features of three anti-CGRP therapeutic antibodies effective in preventing migraine: a comparative pre-clinical case study of ALD403, LY-2951742, TEV-48125. 2016. http://www.alderbio.com/wp-content/uploads/2016/09/EU-Glasgow-2016-ALD403-Comparative-Poster-Latham-v2a-2016-09-08-FINAL2.pdf. Accessed November 9, 2019.
- Vu T, Ma P, Chen JS, et al. Pharmacokinetic-Pharmacodynamic Relationship of Erenumab (AMG 334) and Capsaicin-Induced Dermal Blood Flow in Healthy and Migraine Subjects. Pharm Res. 2017;34(9):1784-1795. Pubmed CrossRef
- Melo-Carrillo A, Strassman AM, Nir RR, et al. Fremanezumab-A Humanized Monoclonal Anti-CGRP Antibody-Inhibits Thinly Myelinated (Adelta) But Not Unmyelinated (C) Meningeal Nociceptors. J Neurosci. 2017;37(44):10587-10596. Pubmed CrossRef
- Schain AJ, Melo-Carrillo A, Stratton J, Strassman AM, Burstein R. CSD-Induced Arterial Dilatation and Plasma Protein Extravasation Are Unaffected by Fremanezumab: Implications for CGRP’s Role in Migraine with Aura. J Neurosci. 2019;39(30):6001-6011. Pubmed CrossRef
- Manoukian R, Sun H, Miller S, Shi D, Chan B, Xu C. Effects of monoclonal antagonist antibodies on calcitonin gene-related peptide receptor function and trafficking. J Headache Pain. 2019;20(1):44. Pubmed CrossRef
- Christensen SL, Petersen S, Kristensen DM, Olesen J, Munro G. Targeting CGRP via receptor antagonism and antibody neutralisation in two distinct rodent models of migraine-like pain. 2019;39(14):1827-1837. Pubmed CrossRef
- Kopruszinski CM, Xie JY, Eyde NM, et al. Prevention of stress- or nitric oxide donor-induced medication overuse headache by a calcitonin gene-related peptide antibody in rodents. 2017;37(6):560-570. Pubmed CrossRef
- Zeller J, Poulsen KT, Sutton JE, et al. CGRP function-blocking antibodies inhibit neurogenic vasodilatation without affecting heart rate or arterial blood pressure in the rat. Br J Pharmacol. 2008;155(7):1093-1103. Pubmed CrossRef
- Ohlsson L, Haanes KA, Kronvall E, Xu C, Snellman J, Edvinsson L. Erenumab (AMG 334), a monoclonal antagonist antibody against the canonical CGRP receptor, does not impair vasodilatory or contractile responses to other vasoactive agents in human isolated cranial arteries. 2019;39(14):1745-1752. Pubmed CrossRef
- Tepper S, Ashina M, Reuter U, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol. 2017;16(6):425-434. Pubmed CrossRef
- Sun H, Dodick DW, Silberstein S, et al. Safety and efficacy of AMG 334 for prevention of episodic migraine: a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Neurol. 2016;15(4):382-390. Pubmed CrossRef
- Dodick DW, Ashina M, Brandes JL, et al. ARISE: A Phase 3 randomized trial of erenumab for episodic migraine. 2018;38(6):1026-1037. Pubmed CrossRef
- Goadsby PJ, Reuter U, Hallstrom Y, et al. A Controlled Trial of Erenumab for Episodic Migraine. N Engl J Med. 2017;377(22):2123-2132. Pubmed CrossRef
- Reuter U, Goadsby PJ, Lanteri-Minet M, et al. Efficacy and tolerability of erenumab in patients with episodic migraine in whom two-to-four previous preventive treatments were unsuccessful: a randomised, double-blind, placebo-controlled, phase 3b study. The Lancet. 2018;392(10161):2280-2287. Pubmed CrossRef
- Reuter U, Goadsby PJ, Lanteri-Minet M, et al. Long-term efficacy and safety of erenumab: Results from 64 weeks of the LIBERTY study. IHC 2019; September 5-8, 2019; Dublin, Ireland.
- Ashina M, Goadsby PJ, Reuter U, et al. Sustained efficacy and long-term safety of erenumab in patients with episodic migraine: 4+ year results of a 5-year, open-label treatment period. IHC 2019; September 5-8, 2019; Dublin, Ireland.
- Ashina M, Goadsby PJ, Reuter U, et al. Long-term safety and tolerability of erenumab: Three-plus year results from a five-year open-label extension study in episodic migraine. 2019;39(11):1455-1464. Pubmed CrossRef
- Ohlsson L, Kronvall E, Stratton J, Edvinsson L. Fremanezumab blocks CGRP induced dilatation in human cerebral, middle meningeal and abdominal arteries. J Headache Pain. 2018;19(1):66. Pubmed CrossRef
- Bigal ME, Edvinsson L, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of chronic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015;14(11):1091-1100. Pubmed CrossRef
- Bigal ME, Dodick DW, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of high-frequency episodic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol. 2015;14(11):1081-1090. Pubmed CrossRef
- Silberstein SD, Dodick DW, Bigal ME, et al. Fremanezumab for the Preventive Treatment of Chronic Migraine. N Engl J Med. 2017;377(22):2113-2122. Pubmed CrossRef
- Dodick DW, Silberstein SD, Bigal ME, et al. Effect of Fremanezumab Compared With Placebo for Prevention of Episodic Migraine: A Randomized Clinical Trial. 2018;319(19):1999-2008. Pubmed CrossRef
- Winner PK, Spierings ELH, Yeung PP, et al. Early Onset of Efficacy With Fremanezumab for the Preventive Treatment of Chronic Migraine. 2019;59(10):1743-1752. Pubmed CrossRef
- Ferrari MD, Diener HC, Ning X, et al. Fremanezumab versus placebo for migraine prevention in patients with documented failure to up to four migraine preventive medication classes (FOCUS): a randomised, double-blind, placebo-controlled, phase 3b trial. 2019;394(10203):1030-1040. Pubmed CrossRef
- Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014;13(9):885-892. Pubmed CrossRef
- Stauffer VL, Dodick DW, Zhang Q, Carter JN, Ailani J, Conley RR. Evaluation of Galcanezumab for the Prevention of Episodic Migraine: The EVOLVE-1 Randomized Clinical Trial. JAMA Neurol. 2018;75(9):1080-1088. Pubmed CrossRef
- Skljarevski V, Matharu M, Millen BA, Ossipov MH, Kim BK, Yang JY. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE-2 Phase 3 randomized controlled clinical trial. 2018;38(8):1442-1454. Pubmed CrossRef
- Ruff DD, Ford JH, Tockhorn-Heidenreich A, et al. Efficacy of Galcanezumab in Patients with Episodic Migraine and a History of Preventive Treatment Failure: Results from Two Global Randomized Clinical Trials. Eur J Neurol. 2019; doi:10.1111/ene.14114. Pubmed CrossRef
- Detke HC, Goadsby PJ, Wang S, Friedman DI, Selzler KJ, Aurora SK. Galcanezumab in chronic migraine: The randomized, double-blind, placebo-controlled REGAIN study. 2018;91(24):e2211-e2221. Pubmed CrossRef
- Camporeale A, Kudrow D, Sides R, et al. A phase 3, long-term, open-label safety study of Galcanezumab in patients with migraine. BMC Neurol. 2018;18(1):188. Pubmed CrossRef
- Dodick DW, Goadsby PJ, Silberstein SD, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014;13(11):1100-1107. Pubmed CrossRef
- Dodick DW, Lipton RB, Silberstein S, et al. Eptinezumab for prevention of chronic migraine: A randomized phase 2b clinical trial. 2019;39(9):1075-1085. Pubmed CrossRef
- Saper JL, R.; Kudrow, D.; Hirman, J.; Dodick, D.; Silberstein, S.; Chakhava, G.; Smith, J. Primary Results of PROMISE-1 (Prevention Of Migraine via Intravenous eptinezumab Safety and Efficacy–1) Trial: a Phase 3, Randomized, Double-blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of Eptinezumab for Prevention of Frequent Episodic Migraines. 2018;90(S20.001).
- Kudrow DL, R.; Silberstein, S.; Cady, R.; Schaeffler, B.; Biondi, D.; Smith, J. Eptinezumab for Prevention of Chronic Migraine: Results of 2 Infusions in the Phase 3 PROMISE-2 (Prevention of Migraine via Intravenous Eptinezumab Safety and Efficacy–2) Trial. 2019;92(P2. 10-006).
- Kudrow D, Berman G, Kassel E, et al. Eptinezumab Treatment for Migraine Prevention Reduces Migraine Disability in Patients with Chronic Migraine: An Analysis from the PREVAIL Open-Label Safety Study. 61st Annual Scientific Meeting American Headache Society; July 11-14, 2019; Philadelphia.
- Silberstein S, Ashina S, Katsarava Z, et al. The Impact of Fremanezumab on Medication Overuse in Patients With Chronic Migraine (P1.10-026). 2019;92(15 Supplement):P1.10-026.
- Schwedt T, Reuter U, Tepper S, et al. Early onset of efficacy with erenumab in patients with episodic and chronic migraine. J Headache Pain. 2018;19(1):92. Pubmed CrossRef
- Goadsby PJ, Dodick DW, Martinez JM, et al. Onset of efficacy and duration of response of galcanezumab for the prevention of episodic migraine: a post-hoc analysis. J Neurol Neurosurg Psychiatry. 2019;90(8):939-944. Pubmed CrossRef
- Silberstein SD, Rapoport AM, Loupe PS, et al. The Effect of Beginning Treatment With Fremanezumab on Headache and Associated Symptoms in the Randomized Phase 2 Study of High Frequency Episodic Migraine: Post-Hoc Analyses on the First 3 Weeks of Treatment. 2019;59(3):383-393. Pubmed CrossRef
- Cohen JM, Dodick DW, Yang R, et al. Fremanezumab as Add-On Treatment for Patients Treated With Other Migraine Preventive Medicines. 2017;57(9):1375-1384. Pubmed CrossRef
- Vo P, Wen S, Martel MJ, Mitsikostas D, Reuter U, Klatt J. Benefit-risk assessment of erenumab and current migraine prophylactic treatments using the likelihood of being helped or harmed. 2019;39(5):608-616. Pubmed CrossRef
- Dong YL, Vegiraju S, Chauhan M, et al. Involvement of calcitonin gene-related peptide in control of human fetoplacental vascular tone. Am J Physiol Heart Circ Physiol. 2004;286(1):H230-239. Pubmed CrossRef
Disclosures
Conflicts of interest: In compliance with the ICMJE uniform disclosure form, all authors declare the following: Dr. Yuan receives honoraria from Supernus Pharmaceuticals, Inc., Dr. Silberstein receives honoraria from Alder Biopharmaceuticals; Allergan, Inc.; Amgen; Avanir Pharmaceuticals, Inc.; Depomed; Dr. Reddy’s Laboratories; eNeura Inc.; electroCore Medical, LLC; Ipsen Biopharmaceuticals; Medscape, LLC; Medtronic, Inc.; Mitsubishi Tanabe Pharma America, Inc.; NINDS; St. Jude Medical; Supernus Pharmaceuticals, Inc.; Teva Pharmaceuticals and Trigemina, Inc.